What Can Model Free Kinetics Tell us About Reaction Mechanisms?
Model free kinetics is based on an isoconversional computational technique that calculates the effective activation energy (E) as a function of the conversion (α) of a chemical reaction, E = f(α).
The variation of E = f(α), is not only of importance for reliable predictions, but also allows one to draw important mechanistic conclusions [1, 2]. For instance, the shape of the activation energy curve indicates directly whether a reaction is simple or more complex. For simple processes, E = f(α) is practically constant (horizontal line). Model free kinetics of thermoanalytically measured reactions, however, rarely gives a constant activation energy.
The fact that a reaction is governed by a constant activation energy does not necessarily mean, however, that it is a single step reaction. Most probably it is a multistep process that is controlled by the rate of the slowest step.
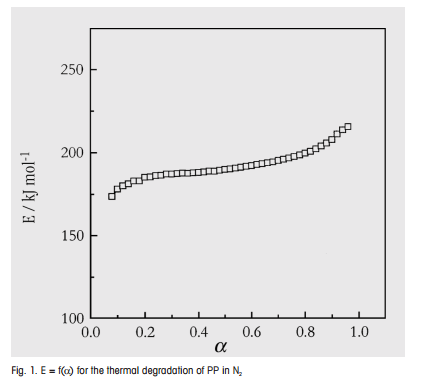
An example of a complex decomposition reaction with almost constant activation energy is the thermal degradation of polypropylene under nitrogen. Figure 1 shows E = f(α) obtained from the model free kinetics of a thermogravimetric study. From the almost constant activation energy of about 180 kJ mol-1, we can conclude that random chain scission is the rate determining step of the degradation reaction.
If E = f(α) is not constant, the process is complex and consists of several reaction steps. Certain complex reactions exhibit a rather typical activation energy dependency E = f(α). For instance, the decomposition reactions of many solid substances are reversible:

The effective activation energy is given by equation (1), [3]:

where E2 is the activation energy of the reverse reaction, λ the heat of absorption, m a constant (0 < m≤ 1),
∆H the heat of reaction, P0 the equilibrium pressure and P the partial pressure of the gaseous reaction product B. From equation 1 it is clear that, as long as the system is not far from equilibrium, the activation energy is temperature dependent because of the temperature dependence of the equilibrium pressure.
If, at the starting temperature, the system is virtually at equilibrium (i.e. P ≈P0) then both the last term of equation 1 and the activation energy are large. With increasing temperature, P0 increases and the system departs from equilibrium, (i.e. P << P0), whereby the last term of equation becomes smaller and decreases to a constant value close to the heat of reaction. The loss of water of crystallization from calcium oxalate monohydrate under non-isothermal conditions is a typical example of this type of kinetic behavior (Figure 2).
If a process involves two (or more) parallel reactions with different activation energies, the contribution of the reaction with the greater activation energy increases with increasing temperature. This is the reason why the experimentally observed activation energy of such processes increases with temperature and therefore also with conversion. An increasing activation energy E = f(α) indicates the occurrence of parallel reactions. An example of this is the kinetics of curing of a DGEBA epoxy resin (diglycidylether of bisphenol A) with phthalic acid anhydride and a tertiary amine as accelerator [4]. The experimentally observed E = f(α) is shown in Figure 3.
From this we can conclude that the curing process consists of two parallel reactions with activation energies of about 20 and 70 kJ mol-1. It is known from the literature [5], that curing is initiated by the reaction of the amine with an epoxy group via the formation of a zwitterion.
The latter reacts with the anhydride to form a carboxyl anion that catalyses the polyaddition. In a competing reaction, the zwitterion reacts with a hydroxyl group of the DGEBA to form an alkoxide anion which promotes the homopolymerization of the DGEBA. In order to test this hypothesis, we measured the activation energy of the pure competing reaction (DGEBA with the amine but without the anhydride) with DSC.
As Figure 3 shows, the polymerization has a practically constant activation energy of about 20 kJ mol-1, which is thus consistent with the value of the competing reaction previously postulated. The low value of E indicates that the process is diffusion controlled. The other reaction with an activation of 70 kJ mol-1 is the polyaddition.

Summary
In summary, it can be said that although the shape of the curve of E = f(α) does not yield any detailed information about the reaction mechanisms, the information is nevertheless adequate for many practical purposes. If, for example, the rate of a reaction that is initially kinetically controlled and then diffusion controlled is to be increased, then the contribution of slow diffusion must be diminished. This can be done by modifying the starting material (e.g. more finely powdered) and optimizing the reaction conditions.
What Can Model Free Kinetics Tell us About Reaction Mechanisms? | Thermal Analysis Application No. UC 104 | Application published in METTLER TOLEDO Thermal Analysis UserCom 10